Targeted EpiEditing
to transform the lives of people
living with severe genetic diseases

Our Approach
Correcting abnormal gene expression exclusively in the cells affected by disease
The Problem
All cell types in the human body require precise control of gene expression to function normally. When this balance is disrupted by a mutation, it can lead to severe genetic diseases with strong unmet medical needs.
The Solution
Regel is pioneering a new class of precision genetic medicine through its Targeted EpiEditing platform, designed to permanently correct gene expression exclusively in the affected cells without altering the genome.
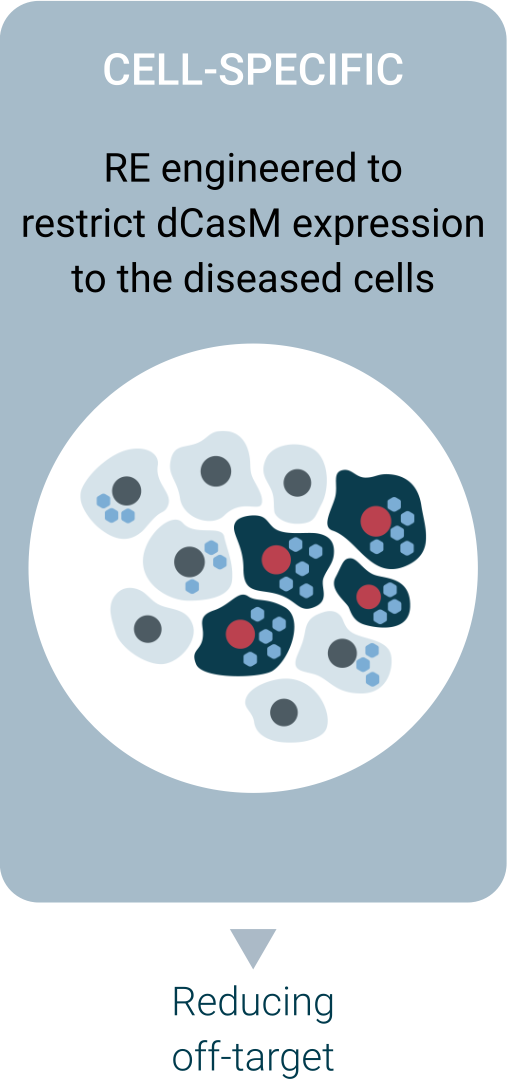
The Platform
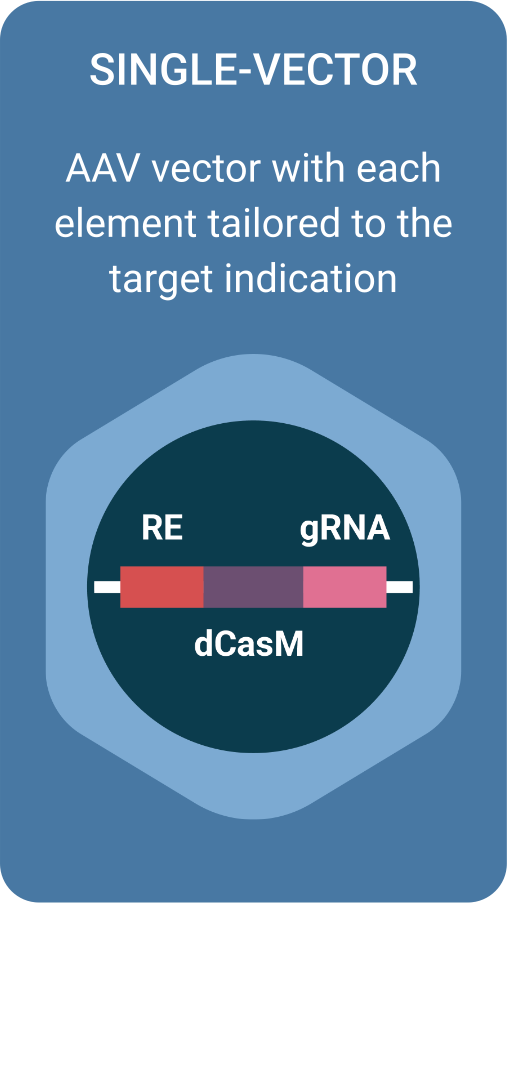
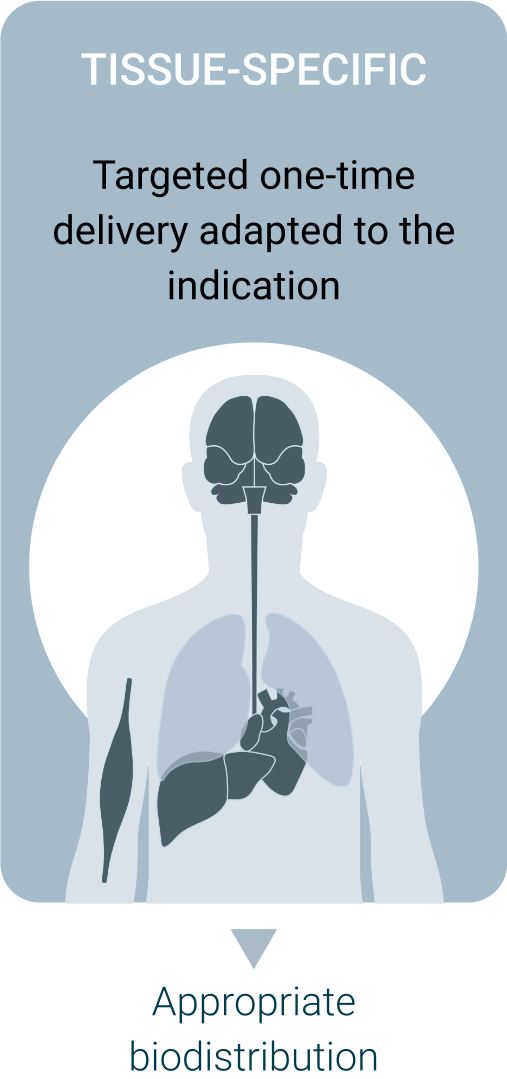
Regel’s technology takes advantage of a deactivated version of CRISPR-Cas9 fused with an epigenetic modulator to permanently correct abnormal gene expression. It is delivered using AAVs equipped with proprietary regulatory elements that confines the intervention to the cells affected by the disease.

Our Pipeline

We are leveraging the Targeted EpiEditing platform for the treatment of severe and life-threatening genetic diseases. Our lead programs for Dravet Syndrome and SCN2A Haploinsufficiency are rapidly advancing towards the clinic.
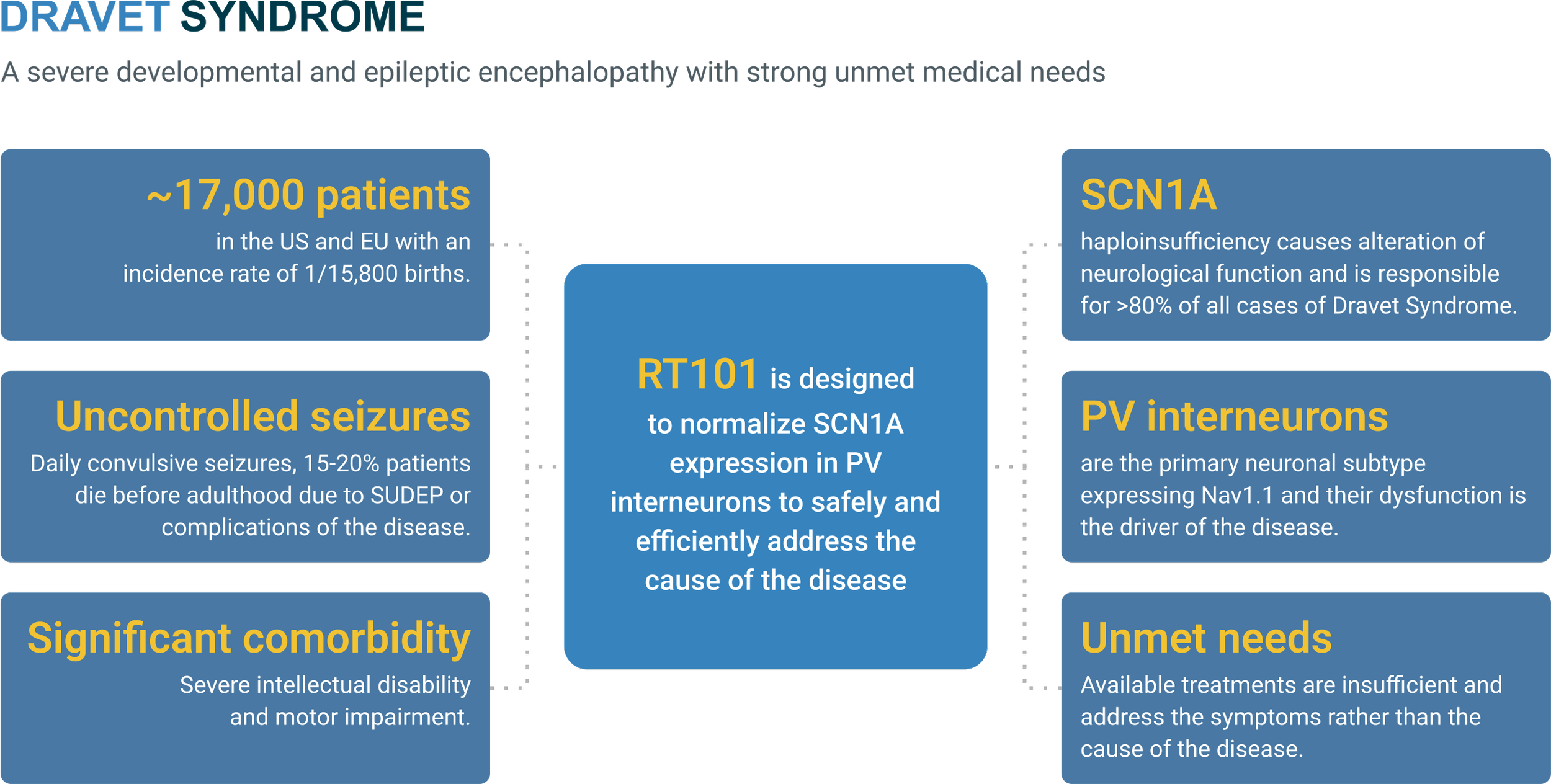
RT101
17,000 patients across US and Europe
suffering from Dravet Syndrome
Preclinical
IND-enabling
Clinical
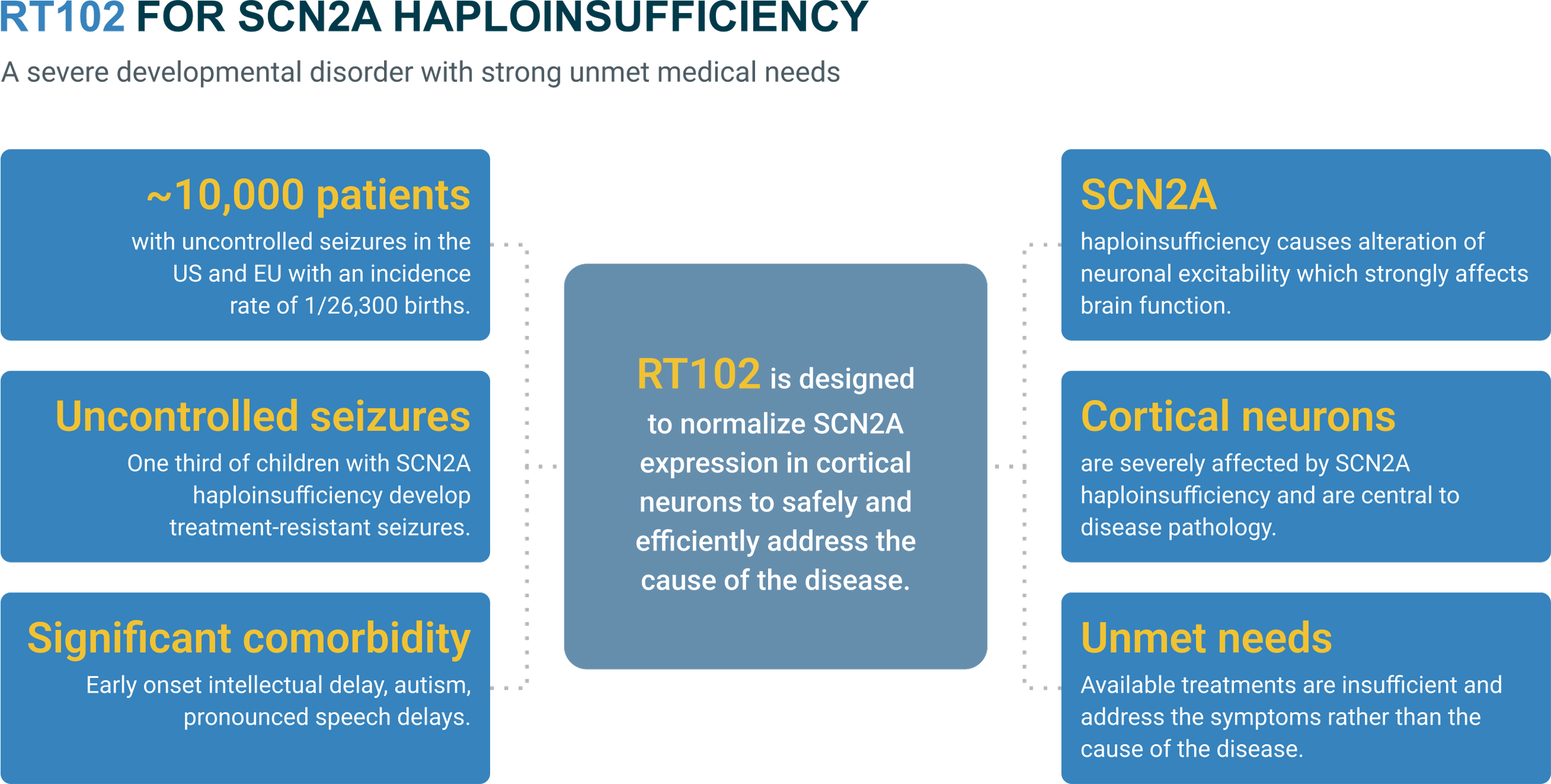
RT102
10,000 patients across US and Europe
suffering from SCN2A Haploinsufficiency
Preclinical
IND-enabling
Clinical

Our Team
Leadership